Dapoxetine Dosage and Price
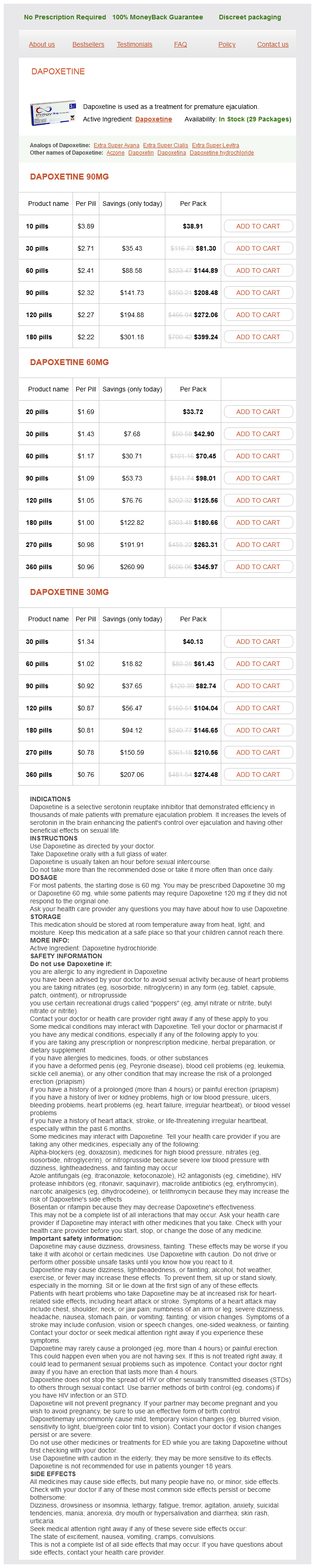
Dapoxetine 90mg
- 10 pills - $38.91
- 30 pills - $81.30
- 60 pills - $144.89
- 90 pills - $208.48
- 120 pills - $272.06
- 180 pills - $399.24
Dapoxetine 60mg
- 20 pills - $33.72
- 30 pills - $42.90
- 60 pills - $70.45
- 90 pills - $98.01
- 120 pills - $125.56
- 180 pills - $180.66
- 270 pills - $263.31
- 360 pills - $345.97
Dapoxetine 30mg
- 30 pills - $40.13
- 60 pills - $61.43
- 90 pills - $82.74
- 120 pills - $104.04
- 180 pills - $146.65
- 270 pills - $210.56
- 360 pills - $274.48
Hypomagnesemia associated with myokymia (quivering of muscles) has been related to a heterozygous loss-of-function mutation (p erectile dysfunction doctors new york 60 mg dapoxetine buy overnight delivery. Hypomagnesemic, hypocalcemic seizures are only transiently responsive and sometimes resistant to parenteral administration of elemental calcium alone. In adult patients with chronic hypomagnesemia, oral preparations of magnesium chloride or glycerophosphate (120 mg thrice daily) may be considered as tolerated; excessively large doses of magnesium may lead to diarrhea and should be avoided. Most neonates with hypermagnesemia are asymptomatic; however, when serum magnesium concentrations are exceptionally high, hypotonia and depression of the central nervous system may be present and when extensive, metabolic bone disease may develop. Hypermagnesemia may also develop in patients with renal insufficiency receiving magnesium-containing antacids. In prematurely delivered neonates, gestational age, birth weight, and rate of postnatal weight gain are important determinants of the postpartum rate of bone mass accrual. After delivery, extremely preterm neonates are unable to maintain the rapid in utero rates of skeletal growth that require synthesis of organic bone matrix and calcium and phosphate deposition into osteoid from the amount of nutrients that can be provided by oral ingestion and gastrointestinal absorption or by parenteral nutrition. Increased maternal parity, male gender, severe systemic disease (bronchopulmonary dysplasia), immobility, and pharmacological agents (glucocorticoids, methylxanthines, such as theophylline, diuretics, such as furosemide) also adversely impact bone formation in these neonates. Approximately 30% (16%40%) of preterm infants with birth weights under 1500 g develop metabolic bone disease within 6 to 16 weeks after birth. Thus intrauterine and postnatal rates of bone turnover in preterm newborns are rapid and persistently elevated through 40 weeks postconceptual age, a conclusion confirmed by bone histomorphometry. The solubility of calcium and phosphate depends not only on their quantities, but also on the forms selected for infusion, that is, calcium chloride, -gluconate, -glycerophosphate, monobasic phosphate, or dibasic phosphate. Using monobasic phosphate and glycerophosphate, it is possible to infuse as much as 86 mg of calcium/kg/dL and 46 mg/kg/dL of phosphate. However, these preparations increase the risk of metabolic acidosis and hypercalciuria. Nevertheless, parenteral nutrition, fortified human milk, and available preterm formulas are unable to provide the amounts of calcium and particularly phosphate that would normally accrue to the fetus in utero. Monitoring of serum levels of calcium, phosphate, creatinine, and alkaline phosphatase and urinary excretion of calcium, phosphate, and creatinine is essential to prevent hypercalcemia and hypercalciuria and nephrocalcinosis. It is also important to avoid hypocalcemia because of the avidity of bone matrix for calcium once remineralization has commenced (hungry bone syndrome). The mutations may be partial gene deletions resulting in decreased synthesis of the triple helix of type I collagen or missense mutations that lead to amino acid substitutions. Lethal osteogenesis imperfecta clinical type 2 may rarely be transmitted as an autosomal dominant trait by a parent who is mosaic for a heterozygous mutation in either collagen-1(I) or -2(I). At the often preterm birth, clinical manifestations of lethal osteogenesis imperfecta include intrauterine growth restriction, long bone deformities and fractures, and a malleable calvarial cortex with large fontanelles; death often occurs in early infancy because of respiratory insufficiency. Radiologically, congenital osteogenesis imperfecta is manifested by short, broad, and "crumpled" long bones, angulation of the tibia, and beading of the ribs. The diagnosis of this illness is most often made in utero; it must be differentiated from achondrogenesis type I, thanatophoric dysplasia, and perinatal hypophosphatasia. Affected patients have impaired urea synthesis because of decreased hepatic uptake of ornithine but are episodically hyperammonemic with increased urinary excretion of the dibasic amino acids; there is extremely low bone mass because of marked protein deprivation and perhaps because of increase in cytokine-induced bone resorption. Administration of citrulline has been reported to increase growth and bone mass in some of these patients. Increased Bone Mass Increased bone mass may be generalized or localized; osteosclerosis refers to thickening of trabecular bone and hyperostosis to increase in cortical bone mass. Infantile osteopetrosis is clinically manifested in affected infants by failure to thrive, delayed development, nasal obstruction, loss of sight, hearing and other cranial nerve functions, and intense bone overgrowth, leading to pancytopenia, hepatosplenomegaly at sites of extramedullary hematopoiesis, and increased susceptibility to infection. Death often occurs within the first several years of life because of sepsis, anemia, or hemorrhage. Additional physical findings in newborns and infants with the most severe form of osteopetrosis include impaired linear growth, macrocephaly, frontal bossing, nystagmus, delayed eruption of primary teeth, and ecchymoses. The radiographic hallmark of infantile osteopetrosis is relatively uniform increase in bone density of the skull, vertebrae, and axial skeleton with thickened cortical and trabecular bone, "Ehrlenmeyer flask" deformities at the distal ends of the long bones in older children, and alternating bands of sclerotic and lucent bone in the iliac wings and metaphyses. In selected subjects, bone marrow transplantation providing osteoclast precursor cells has been effective in arresting progression of this disorder, albeit often with substantial residual deficits. Bone mineralization may be excessive because of increase in the rate of mineral deposition or decrease in the rate of resorption of the mineral phase of bone. Ectopic calcification of extraskeletal tissues may occur when local calcium and phosphate levels are high, whereas extraskeletal ossification may ensue when the regulation of skeletal bone formation is deranged. Rickets Rickets in children is a disorder of the cartilaginous growth plate and the failure to deposit sufficient bone mineral (hydroxyapatite) to achieve necessary bone strength enabling upright weight-bearing and ambulation because of deficiency of calcium, phosphate, vitamin D, or one of many cofactors that ensure appropriate assimilation and utilization of these materials. Thus rickets is a childhood disorder of decreased mineralization of cartilage and primary bone spongiosa of the growth plates at the ends of the long bones and of bone matrix collagen type I below the growth plate that leads to impaired skeletal strength and linear growth and bony deformations (craniotabes, metaphyseal flaring, genu valgum, genu varum), and increased fracture risk. In the adult, deficiencies of vitamin D, calcium, or phosphate result in osteomalacia, the failure to mineralize the cortex and trabeculae of bone. During endochondral bone formation in growing children, cartilage matrix is elaborated and subsequently mineralized. When endochondral matrix is not fully mineralized, cartilage accumulates, there is thickening of the growth plate and disorganization of the chondrocytes; the ends of the long bones (particularly those that are weight-bearing) are distorted resulting in rachitic deformities. During intervals of calcium and/or phosphate deprivation, the actively growing, weight-bearing child with open cartilaginous growth plates develops rickets and osteomalacia, whereas under similar circumstances adults develop only osteomalacia during remodeling as unmineralized bone matrix accumulates. Thus rickets is the expression of defective endochondral mineralization at the growth plate and osteomalacia is the failure of mineralization of bone cortex and trabeculae. Minireview: fibroblast growth factor 23 in phosphate homeostasis and bone metabolism. The Thacher radiographic scoring system for assessment of the severity of rickets provides a reasonably objective and useful method for determining the extent of rickets and its response to treatment.
Presbyopia · With increasing age erectile dysfunction drugs stendra cheap dapoxetine 90 mg buy on-line, crystalline lens loses its elasticity and power of accommodation for near vision (reading). These changes are more evident by the age of 40 to 45 years and the condition is called presbyopia. Meibomian/tarsal glands: these are 2025 long sebaceous glands that are embedded in the tarsal plates. Gland of Zeiss (sebaceous glands of eyelashes): these are small, modified sebaceous glands. Gland of Moll (apocrine glands of eyelashes): these are modified sweat glands present near the free edges of eyelids. Glands of Wolfring are located on inner surface of upper eyelid, whereas glands of Krause open into the fornix of lacrimal sac. Neet Clinical Correlation · Chalazion: It is an inflammation and blockade of tarsal gland. Usually, viral conjunctivitis does not need any treatment other than warm compression and artificial tears. Membranous labyrinth: It consists of duct of cochlea, semicircular ducts, ductus reuniens, utricle, saccule, and endolymphatic duct and sac. Middle Ear · Middle ear is a small space in the petrous part of temporal bone · Middle ear is lined by ciliated columnar or cuboidal epithelium. Innermost mucous layer · Fibrous layer has superficial radiating fibers and deep circular collagen fibers. Fenestra cochleae/round window that is closed by a membrane called secondary tympanic membrane. Semicircular Canals · There are three bony semicircular canals: Superior, posterior, and lateral. Semicircular Ducts · Three semicircular ducts lie within the bony semicircular canals (superior, posterior, and lateral). These consist of thin fibrous sheet lined with simple squamous or cuboidal epithelium. Supporting Cells · Organ of Corti has supporting cells such as inner and outer rod cells, phalangeal cells of Dieters, cells of Hensen, and cells of Claudius. Structure · A macula has two parts: Sensory neuroepithelium and otolithic membrane. They are multitude of small cylindrical and hexagonally shaped bodies with pointed ends. Supporting/sustentacular cells · Supporting cells are elongated and may be shaped like hour glasses (narrow in the middle and wide at each end). Hair cells · Apical surface of hair cells has two types of hairs: Single large kinocilium and numerous stereocilia (large microvilli). One of the genes removed by the deletion in these cases, esterase D, then acted as a polymorphic genetic linkage marker for further studies. The first hit, including the germline one, is usually a point mutation, with the second lost by large deletions often of the chromosome arm. It is the autosomal dominant disorders that have been of most interest as they are likely to represent the inheritance of a mutated copy of a tumour suppressor gene, which also predisposes the individual to common cancers. Although these conditions are generally uncommon, the tumour suppressor genes involved o en play a fundamental role in the causation and initiation of sporadic cancers, which a ect 3540% of humans in the developed world in their lifetime. If left without intervention this leads to the almost inevitable development of a colorectal cancer by 60 years of age. The condition may also be associated with osteomas, desmoid tumours and epidermal cysts, as well as an increased risk of other malignancies such as duodenal cancer, hepatoblastoma, glioma, medulloblastoma and thyroid cancer. Patients with mutations in the 5 exons 25 (early part of the gene reading frame) had a very mild clinical picture with late onset of polyps, whereas those with mutations from exon 9 to codon 1450 of exon 15 had a classical disease course with nearly all patients manifesting the typical congenital retinal pigmentation [11]. However, those with mutations beyond codon 1450 showed typical Gardner syndrome features (osteomas, cysts and desmoid disease) without congenital hypertrophy of the retinal pigment epithelium, but also had milder polyp disease [11]. Gorlin syndrome is characterized by the development of multiple jaw keratocysts and basal cell carcinomas as well as a 25% risk of childhood medulloblastoma [18]. Additional important dominant conditions include PeutzJeghers [23] and juvenile polyposis, [24] both of which have a substantial risk of gastrointestinal tract malignancy. All of these cancer-predisposing syndromes have bene ted from the identi cation of the causative gene and the targeted use of screening which in most of the conditions improves life expectancy [25]. In addition to family history, cancers are more likely to be hereditary if they are early onset, bilateral, multifocal or are associated with other related primaries (such as breast with ovarian cancer or colorectal and endometrial). Risk-reducing surgery can reduce risks of both cancers by more than 90%, with oophorectomy conferring protection against ovarian cancer and breast cancer [3739]. Recently survival advantages have been published for those undergoing contralateral mastectomy a er breast cancer [40] as well as for primary prevention [41]. Most cancers require a number of genetic mutations in a progenitor cell before an invasive tumour results. Virtually none will be caused solely by the loss of two copies of a single tumour suppressor gene as in retinoblastoma, and the number of driver mutations probably varies between 4 and 10 (many more are present on genome sequencing but most are bystanders). A combination of loss of function of tumour suppressor genes and activation of oncogenes is normally involved. Adenocarcinomas are more likely than carcinomas of squamous epithelium to have a strong hereditary component with 416% of all breast, ovarian and colon 22 Genetics and hereditary cancer syndromes, genetic polymorphisms and cancer Table 2. However, since 2013 many commercial companies and health services have moved to testing panels of known cancer-predisposing genes, which may not even target the organs indicated from the family history [47,48], for example testing colorectal cancer genes in a breast cancer family. A further 16 had what were classi ed as pathogenic mutations in the extended panel of 42 genes. Inheriting a germline mutation confers about a 3080% lifetime colorectal cancer risk without colonoscopy [50]. However, mutations also enhance risks of endometrial, ovarian, gastric and upper urinary tract cancers [51].
Osteogenesis imperfecta clinical type 2 can be identified prenatally by fetal ultrasonography; other types might be determined prenatally by analysis of collagen synthesized by cells cultured from chorionic villus biopsies and by genetic analyses erectile dysfunction treatment home veda buy dapoxetine 60 mg mastercard. Other clinical findings of note in patients with osteogenesis imperfecta include: progressive hearing impairment-present in 40% to 60% of subjects, joint hypermobility in perhaps 60% to 70% of patients that may lead to joint dislocation and/or tendon rupture, and craniocervical junction insults. Cranial-cervical deformities occur in approximately 30% of patients with osteogenesis imperfecta and may be categorized as: basilar invagination, basilar impression, and platybasia- the most common of these complications. Symptoms/signs of cranial involvement are headaches on movement, coughing or sneezing; trigeminal neuralgia; weakness of arms/legs; and difficulties with balance. Screening of subjects with osteogenesis imperfecta for these complications is essential. Keys to the prevention of children and adolescents from developing low bone mass is the provision of a diet adequate in calcium and vitamin D, encouragement of weight-bearing activities and exercise, and the avoidance of exposure to agents that may impede normal accrual of bone mineral. After assessment and when necessary and appropriate, interventions may include treatment of an accompanying underlying systemic disease or endocrinopathy that may be of pathogenetic significance in the development of low bone mass, such as elimination, such as elimination or reduction in glucocorticoid dose in an asthmatic child. Therapeutic agents used in the treatment of patients with low bone mass increase skeletal mass either by inhibiting bone resorption (antiresorptive or antiremodeling drugs) or by stimulating bone formation (anabolic agents). Although approved for treatment of osteoporosis in adults, denosumab and romosozumab await further development and evaluation in children with osteoporosis of varied pathogenesis. Infants/children/adolescents with osteogenesis imperfecta require care by a team of experienced endocrinologists, orthopedic surgeons, and physiatrists and their corresponding healthcare associates. Rehabilitative services and physical therapy to improve muscle strength and mobility with the restraints of bone fragility are encouraged as is protected exercise-such as walking and swimming. Patients with primary osteogenesis imperfecta (and secondary forms of osteoporosis) with documented vertebral and/or long bone fractures have been treated most often with bisphosphonates. Bisphosphonates that contain nitrogen as a constituent in one of the side chains are substantially more potent than are the "simple" bisphosphonates. Bisphosphonates chelate (bind) the calcium ions of hydroxyapatite and thus are targeted to bone; within the resorption lacuna beneath an osteoclast, bisphosphonates dissociate from hydroxyapatite as the pH is lowered by osteoclast secretion of H+ and are then endocytosed into the interior of the osteoclast. This enzyme is essential for synthesis of cholesterol through the mevalonate pathway; its inhibition prevents prenylation of proteins, a posttranslational modification that enables prenylated proteins to interact with proteins and to bind to cell membranes. The biological activity of a bisphosphonate on osteoclast function is observed immediately after its administration as serum calcium concentrations decline rapidly; indeed, this rapid effect has been used in the treatment of hypercalcemic patients. The effects of bisphosphonates on bone last long after the agent has been discontinued (the "residence time") enabling some compounds to be given as infrequently as once yearly or halfyearly. Histomorphometric analysis has revealed that bisphosphonate-mediated inhibition of osteoclast stimulated bone resorption increase bone mineralization by decreasing the number of resorption cavities and thus the remodeling space, preserving cancellous (trabecular) bone architecture, and decreasing porosity of cortical bone. Bisphosphonates have been useful in improving mineralization in children with osteogenesis imperfecta and related syndromes, as well as in those with glucocorticoid-induced osteoporosis and those in whom mobility is limited, such as muscular dystrophies and cerebral palsy. Discontinuation of bisphosphonate administration may lead to increase in fracture risk between weaker new bone and stronger treated bone. To avoid medicationrelated systemic effects, such as fever, malaise, and myalgia, administration of an analgesic and antipyretic agent for 48 to 72 hours after administration of the bisphosphonate is often used. Serious side effects of bisphosphonates are unusual but include iritis, atypical femoral fracture caused by decreased bone remodeling, and osteonecrosis of the jaw, the latter complication being unusual in children. Abnormalities of Mineral Homeostasis in the Newborn, Infant, Child, and Adolescent 785 Side effects of bisphosphonates have been both acute (fever, myalgia, abdominal pain, vomiting, hypocalcemia) and chronic (inflammatory disorders of the eye, osteonecrosis of the jaw in the elderly, and induced "osteopetrosis") but have not generally occurred in the pediatric population. Treatment with bisphosphonates several years before conception does not appear to have an adverse effect on fetal outcome, but treatment during pregnancy is contraindicated because of possible toxicity. When administered intermittently in small amounts, both agents preferentially accelerate the rates of bone remodeling and of bone formation relative to that of bone resorption by direct effects upon osteoblast differentiation, maturation, and longevity. Although osteosarcoma has been observed in mice receiving very high doses of these agents, no malignant disorders have been recorded in adults receiving either agent. Infants as young as 2 months of age have safely tolerated 4-hour intravenous infusions of pamidronate (0. During 2 to 4 years of intravenous pamidronate administration, increase in vertebral (trabecular) bone mass and size are accompanied by decline in the extent of vertebral compression and fewer compressed vertebrae than in untreated patients. Fibrodysplastic lesions are initially silent whereas osteoclasts at the periphery of the lesions actively compress and thin bone cortices, ultimately resulting in bone pain and pathological fractures of the long bones, particularly the proximal femoral metaphyses. Within the skull base and facial bones, expansion of fibrous dysplastic lesions leads to disfiguration and compression of cranial nerves. Radiographically, the fibrodysplastic lesion is viewed as a "cyst-like" medullary structure with a "ground-glass" consistency without a trabecular pattern. The clinical manifestations of fibrous dysplasia depend on the sites and extent of bone involvement and associated endocrinopathies. In addition to managing the multiple endocrinopathies and organ defects, attention must be paid to the osseous lesions. Fractures are repaired by standard techniques, including intramedullary nailing when indicated; occasionally it may be feasible to evacuate a fibrodysplastic lesion surgically and to fill the cavity with bone grafts. In patients with polyostotic fibrous dysplasia without or associated with the McCune-Albright syndrome, administration of the oral bisphosphonate alendronate did not alter the radiographic appearance of the skeletal lesions, ameliorate bone pain, or improve function. Cessation of denosumab was marked by rebound increase in values of bone turnover markers and hypercalcemia. High Bone Mass Abnormally increased bone mass is the consequence of disruption of the normal equilibrium between the coupled processes of bone formation and resorption. Specifically, increase in cortical bone width is termed hyperostosis; thickening of trabecular bone is termed osteosclerosis. Failure of osteoclast-mediated bone resorption leads to osteopetrosis that may be associated with a large number of poorly functioning osteoclasts ("osteoclast-rich" osteopetrosis) or with a normal number or paucity of osteoclasts ("osteoclastpoor" osteopetrosis). Autosomal dominant type 1 osteopetrosis is not related to an error in osteoclast differentiation or function and defective bone resorption but rather is associated with increased osteoblastogenesis resulting in enhanced formation and mineralization of bone.